


{kind=link}
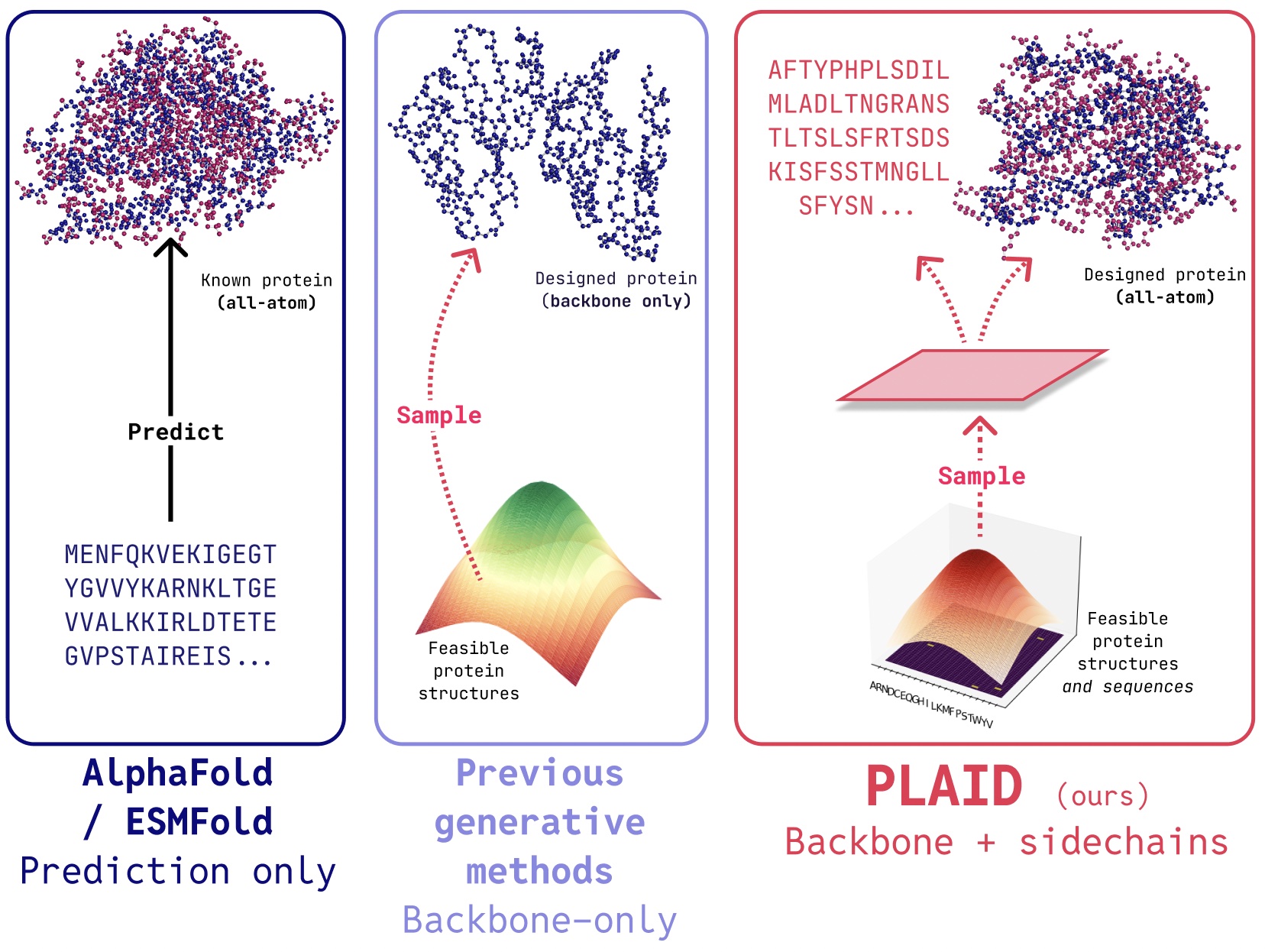
TARTÁN es un modelo generativo multimodal que genera simultáneamente la secuencia de proteína 1D y la estructura 3D, al aprender el espacio latente de los modelos de plegamiento de proteínas.
La adjudicación de la 2024 Premio Nobel Alfafold2 marca un momento importante de reconocimiento para el papel de IA en la biología. ¿Qué viene después después del plegamiento de proteínas?
En TARTÁNdesarrollamos un método que aprende a probar desde el espacio latente de los modelos de plegamiento de proteínas a generar Nuevas proteínas. Puede aceptar Función de composición y indicaciones de organismoy puede ser capacitado en bases de datos de secuenciaque son 2-4 órdenes de magnitud mayores que las bases de datos de estructura. A diferencia de muchos modelos generativos de estructura de proteínas anteriores, el cuadro de cuadros aborda la configuración de problemas de co-generación multimodal: generando simultáneamente la secuencia discreta y las coordenadas estructurales continuas de todo el átomo.
Desde la predicción de la estructura hasta el diseño de medicamentos del mundo actual
Aunque los trabajos recientes demuestran promesa para la capacidad de los modelos de difusión para generar proteínas, todavía existen limitaciones de modelos anteriores que los hacen poco prácticos para aplicaciones del mundo actual, como:
- Generación de átomos: Muchos modelos generativos existentes solo producen átomos de columna vertebral. Para producir la estructura de todos los átomos y colocar los átomos de la cadena lateral, necesitamos conocer la secuencia. Esto crea un problema de generación multimodal que requiere una generación simultánea de modalidades discretas y continuas.
- Especificidad del organismo: Las proteínas biológicas destinadas al uso humano deben ser humanizadopara evitar ser destruido por el sistema inmune humano.
- Especificación de management: El descubrimiento de fármacos y ponerlo en manos de los pacientes es un proceso complejo. ¿Cómo podemos especificar estas limitaciones complejas? Por ejemplo, incluso después de abordar la biología, puede decidir que las tabletas son más fáciles de transportar que los viales, agregando una nueva restricción a la solucabilidad.
Generar proteínas “útiles”
Simplemente generar proteínas no es tan útil como controlador la generación para obtener útil proteínas. ¿Cómo podría ser una interfaz para esto?

Para inspiración, consideremos cómo controlaríamos la generación de imágenes a través de indicaciones textuales de composición (ejemplo de Liu et al., 2022).
En cuadros, reflejamos esta interfaz para especificación de management. El objetivo closing es controlar la generación por completo a través de una interfaz textual, pero aquí consideramos restricciones compositivas para dos ejes como una prueba de concepto: función y organismo:
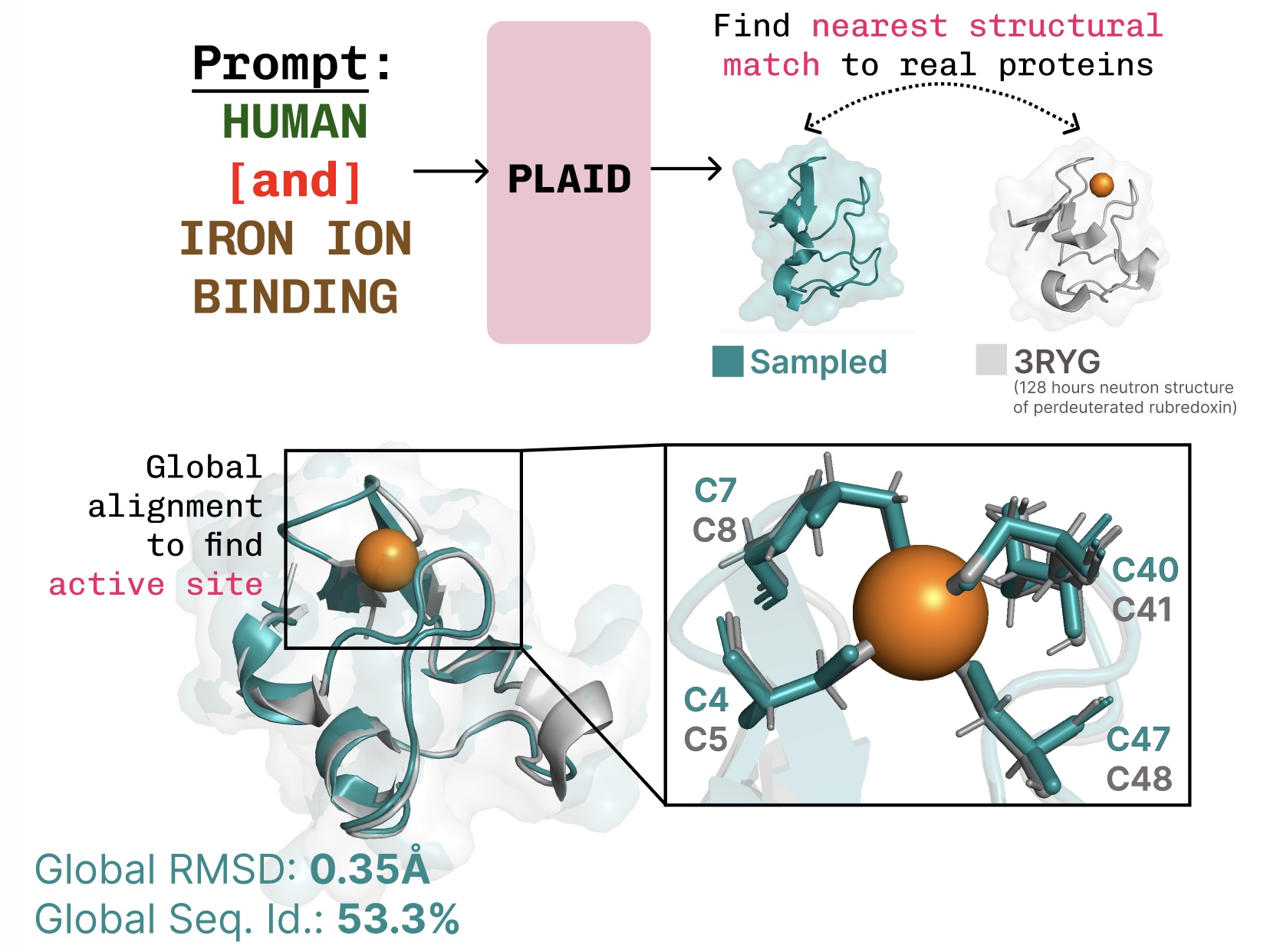
Aprendiendo la conexión de la estructura-estructura-secuencia. El cuadrado aprende el tetraédrico cisteína-FE2+/Fe3+ Patrón de coordinación a menudo que se encuentra en las metaloproteínas, al tiempo que mantiene una alta diversidad de nivel de secuencia.
Entrenamiento utilizando datos de capacitación de solo secuencia
¡Otro aspecto importante del modelo a cuadros es que solo requerimos secuencias para entrenar el modelo generativo! Los modelos generativos aprenden la distribución de datos definida por sus datos de entrenamiento, y las bases de datos de secuencia son considerablemente más grandes que las estructurales, ya que las secuencias son mucho más baratas de obtener que la estructura experimental.
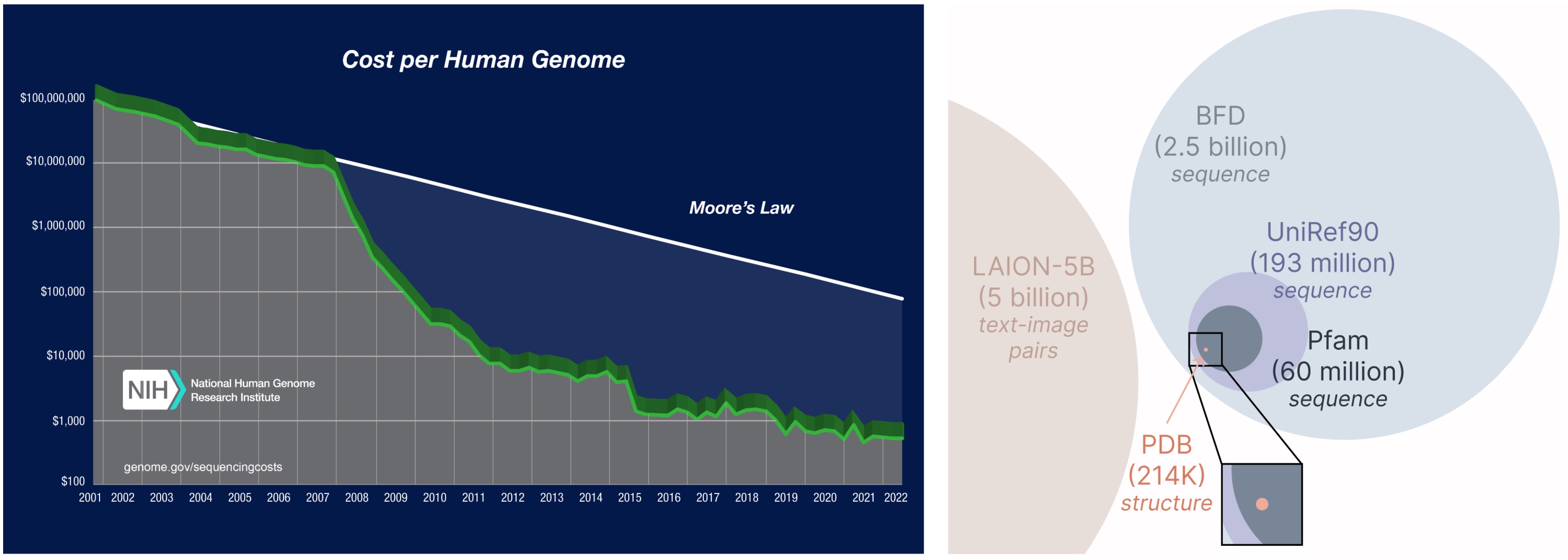
Aprender de una base de datos más grande y más amplia. El costo de obtener secuencias de proteínas es mucho más bajo que la estructura de caracterización experimental, y las bases de datos de secuencia son 2-4 órdenes de magnitud mayores que las estructurales.
¿Cómo funciona?
La razón por la que podemos entrenar el modelo generativo para generar la estructura mediante el uso de datos de secuencia es aprendiendo un modelo de difusión sobre el espacio latente de un modelo de plegado de proteínas. Luego, durante la inferencia, después de muestrear desde este espacio latente de proteínas válidas, podemos tomar pesas congeladas Desde el modelo de plegamiento de proteínas hasta la estructura de decodificación. Aquí, usamos Esmfoldun sucesor del modelo Alfafold2 que reemplaza un paso de recuperación con un modelo de lenguaje de proteína.
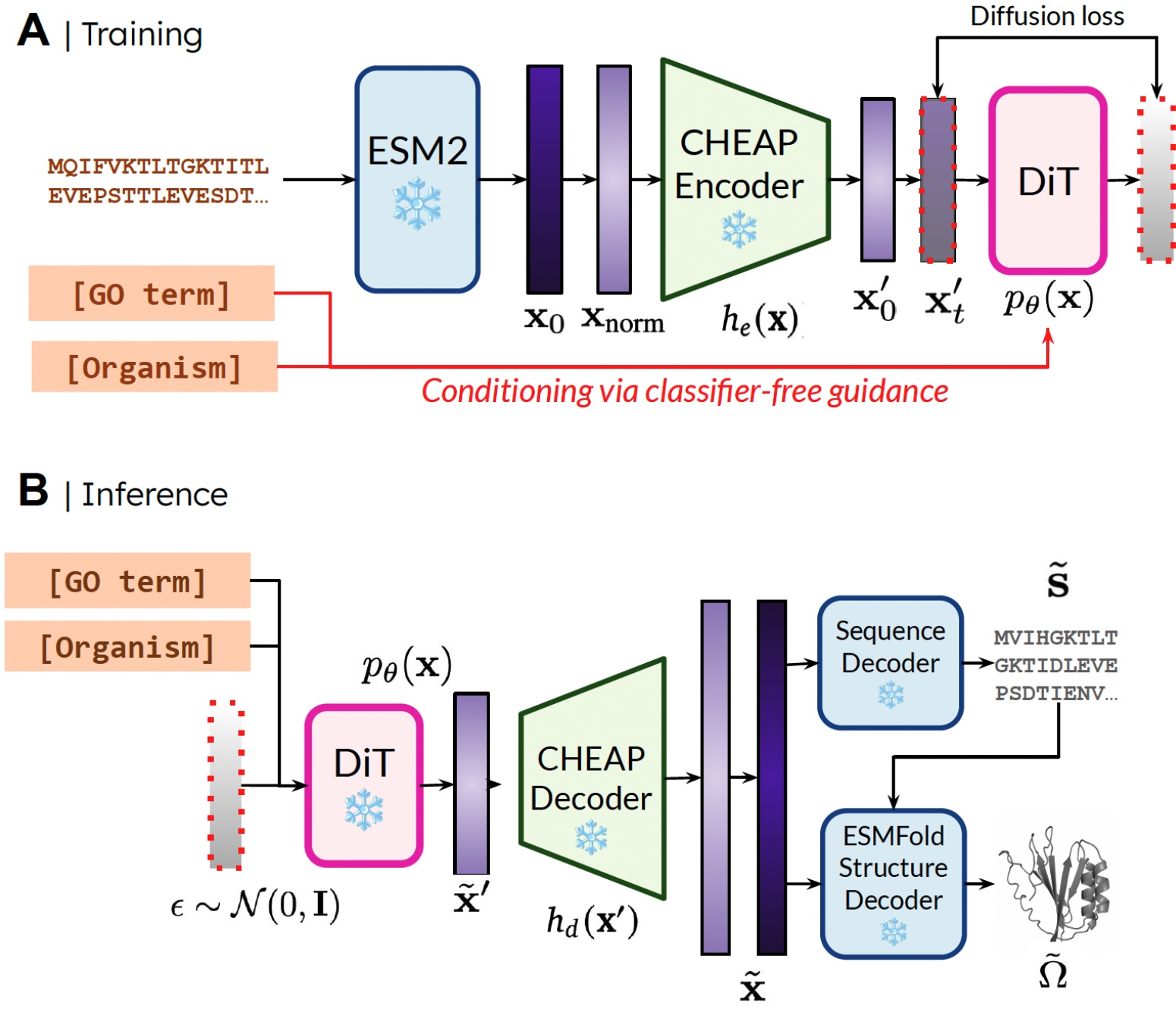
Nuestro método. Durante el entrenamiento, solo se necesitan secuencias para obtener la incrustación; Durante la inferencia, podemos decodificar la secuencia y la estructura de la incrustación muestreada. ❄️ denota pesas congeladas.
De esta manera, podemos utilizar la información de comprensión estructural en los pesos de los modelos de plegamiento de proteínas previos a la aparición para la tarea de diseño de proteínas. Esto es análogo a cómo los modelos de acción-lenguaje de la visión (VLA) en robótica hacen uso de los antecedentes contenidos en los modelos de lenguaje de visión (VLMS) capacitados en datos a escala de Web para proporcionar percepción y razonamiento y comprensión de información.
Comprimiendo el espacio latente de los modelos de plegamiento de proteínas
Una pequeña arruga al aplicar directamente este método es que el espacio latente de ESMfold, de hecho, el espacio latente de muchos modelos basados en transformadores, requiere mucha regularización. Este espacio también es muy grande, por lo que aprender esta incrustación termina mapeo a la síntesis de imágenes de alta resolución.
Para abordar esto, también proponemos BARATO (Adaptaciones de proteínas de enviornment comprimida de proteínas)donde aprendemos un modelo de compresión para el incrustación articular de secuencia y estructura de proteínas.
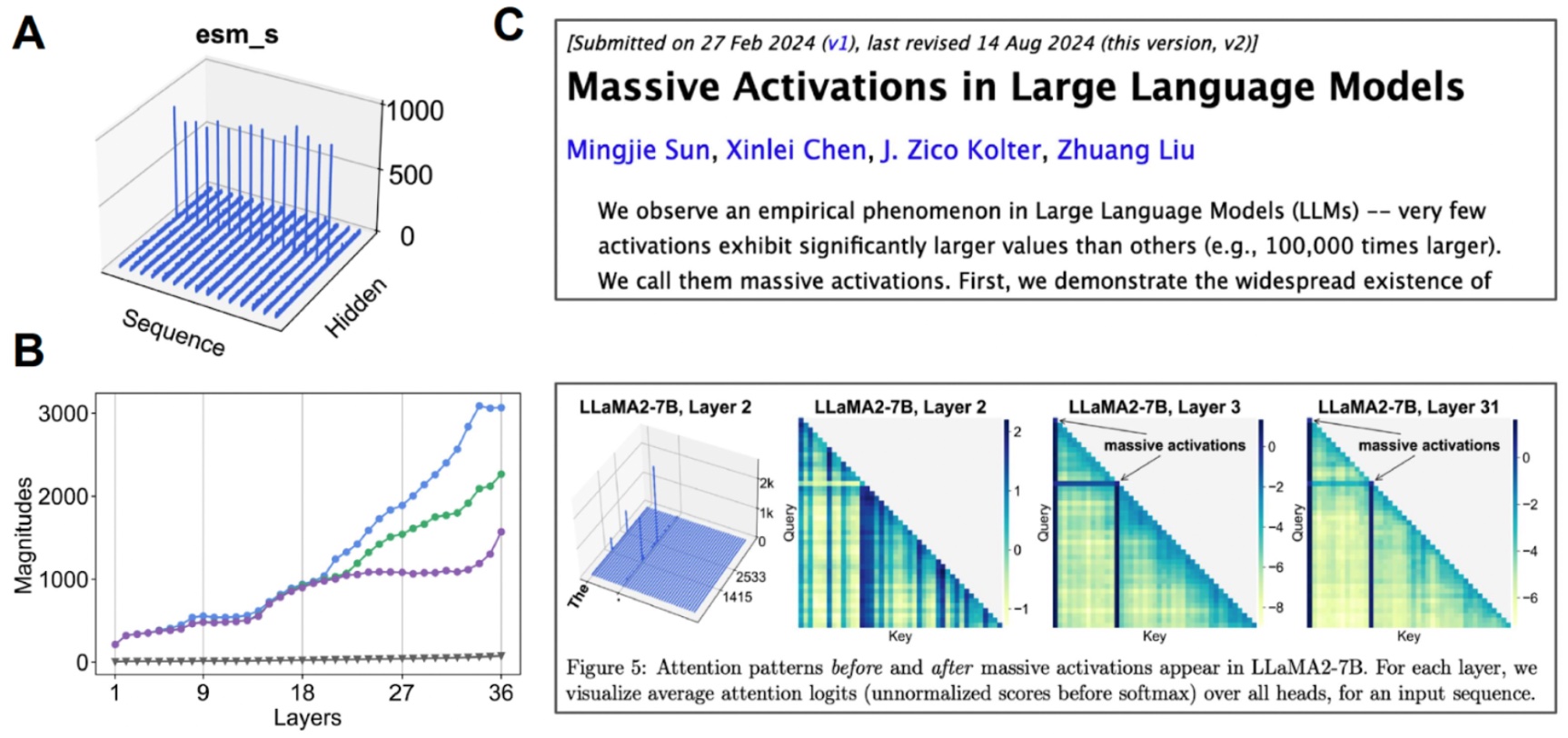
Investigando el espacio latente. (A) Cuando visualizamos el valor medio para cada canal, algunos canales exhiben “activaciones masivas”. (B) Si comenzamos a examinar las activaciones de Prime 3 en comparación con el valor medio (gris), encontramos que esto sucede en muchas capas. (C) También se han observado activaciones masivas para otros modelos basados en transformadores.
Encontramos que este espacio latente es realmente altamente compresible. Al hacer un poco de interpretabilidad mecanicista para comprender mejor el modelo base con el que estamos trabajando, pudimos crear un modelo generativo de proteínas de átomo.
¿Qué sigue?
Aunque examinamos el caso de la secuencia de proteínas y la generación de estructuras en este trabajo, podemos adaptar este método para realizar una generación multimodal para cualquier modalidad donde haya un predictor de una modalidad más abundante a una menos abundante. Como los predictores de secuencia a estructura para las proteínas están comenzando a abordar sistemas cada vez más complejos (por ejemplo, AlphaFold3 también puede predecir proteínas en complejos con ácidos nucleicos y ligandos moleculares), es fácil imaginar realizar una generación multimodal sobre sistemas más complejos utilizando el mismo método. Si está interesado en colaborar para extender nuestro método o probar nuestro método en el laboratorio húmedo, ¡comuníquese!
Enlaces adicionales
Si ha encontrado nuestros documentos útiles en su investigación, considere usar el siguiente bibtex para cuadros y baratos:
@article{lu2024generating,
title={Producing All-Atom Protein Construction from Sequence-Solely Coaching Information},
writer={Lu, Amy X and Yan, Wilson and Robinson, Sarah A and Yang, Kevin Okay and Gligorijevic, Vladimir and Cho, Kyunghyun and Bonneau, Richard and Abbeel, Pieter and Frey, Nathan},
journal={bioRxiv},
pages={2024--12},
12 months={2024},
writer={Chilly Spring Harbor Laboratory}
}
@article{lu2024tokenized,
title={Tokenized and Steady Embedding Compressions of Protein Sequence and Construction},
writer={Lu, Amy X and Yan, Wilson and Yang, Kevin Okay and Gligorijevic, Vladimir and Cho, Kyunghyun and Abbeel, Pieter and Bonneau, Richard and Frey, Nathan},
journal={bioRxiv},
pages={2024--08},
12 months={2024},
writer={Chilly Spring Harbor Laboratory}
}
También puede pagar nuestras preimpresiones (TARTÁN, BARATO) y CodeBases (TARTÁN, BARATO).
¡Alguna diversión de generación de proteínas de bonificación!
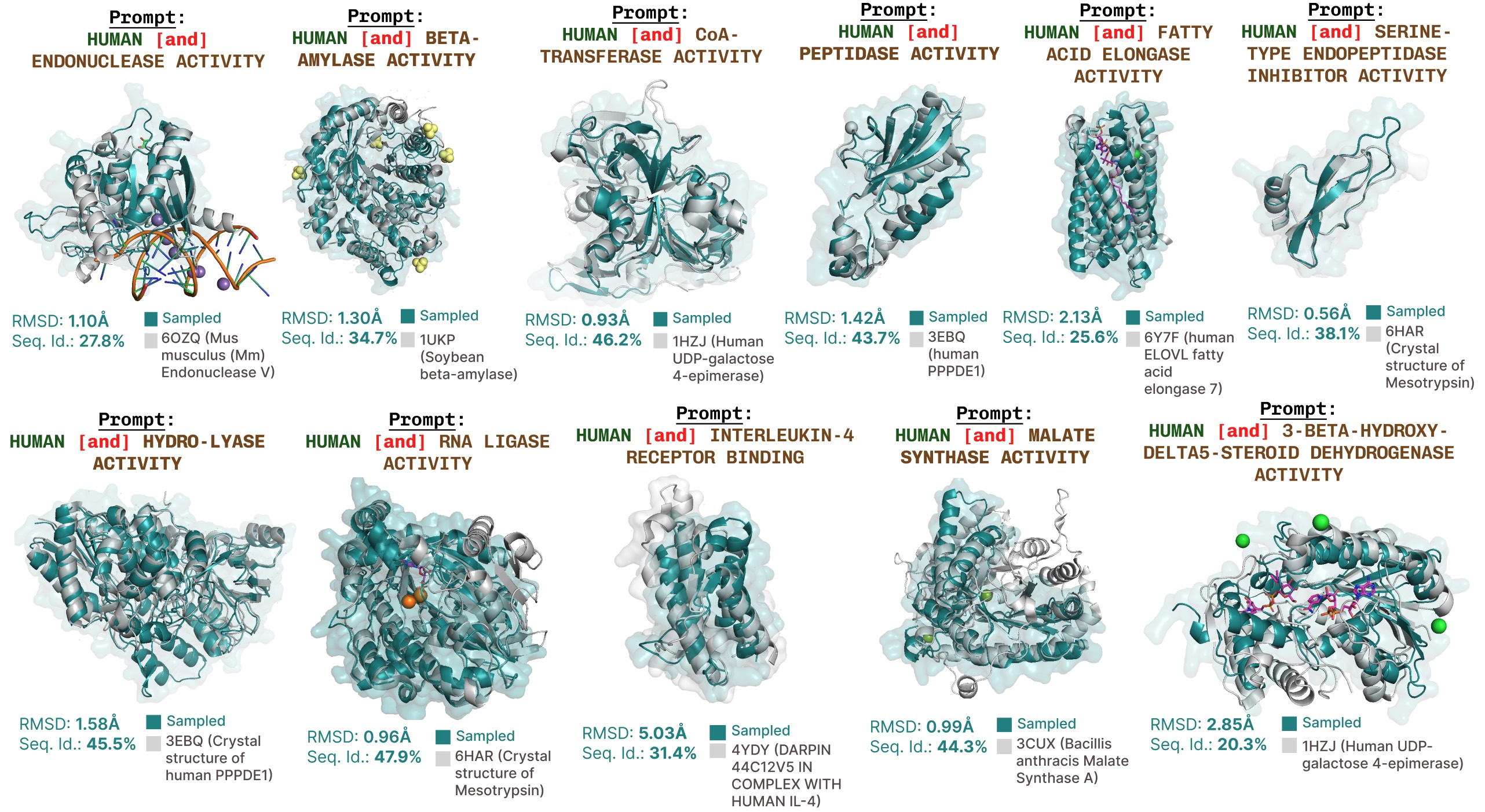
Generaciones adicionales prometidas por funciones con cuadros.
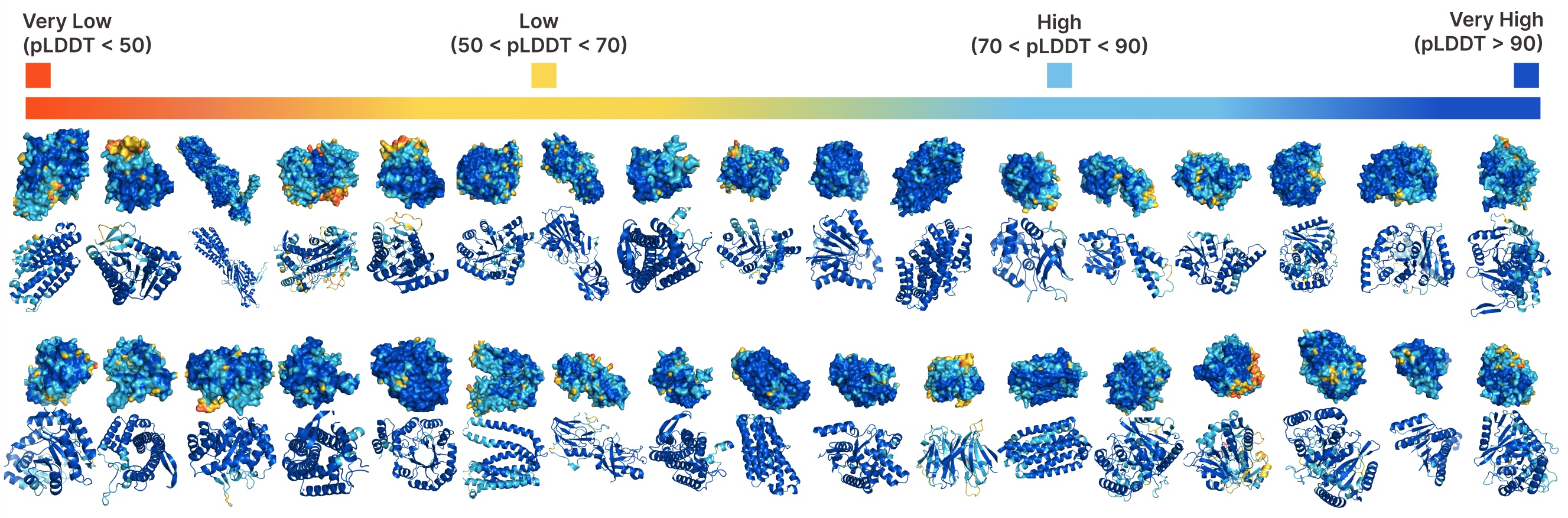
Generación incondicional con cuadros.
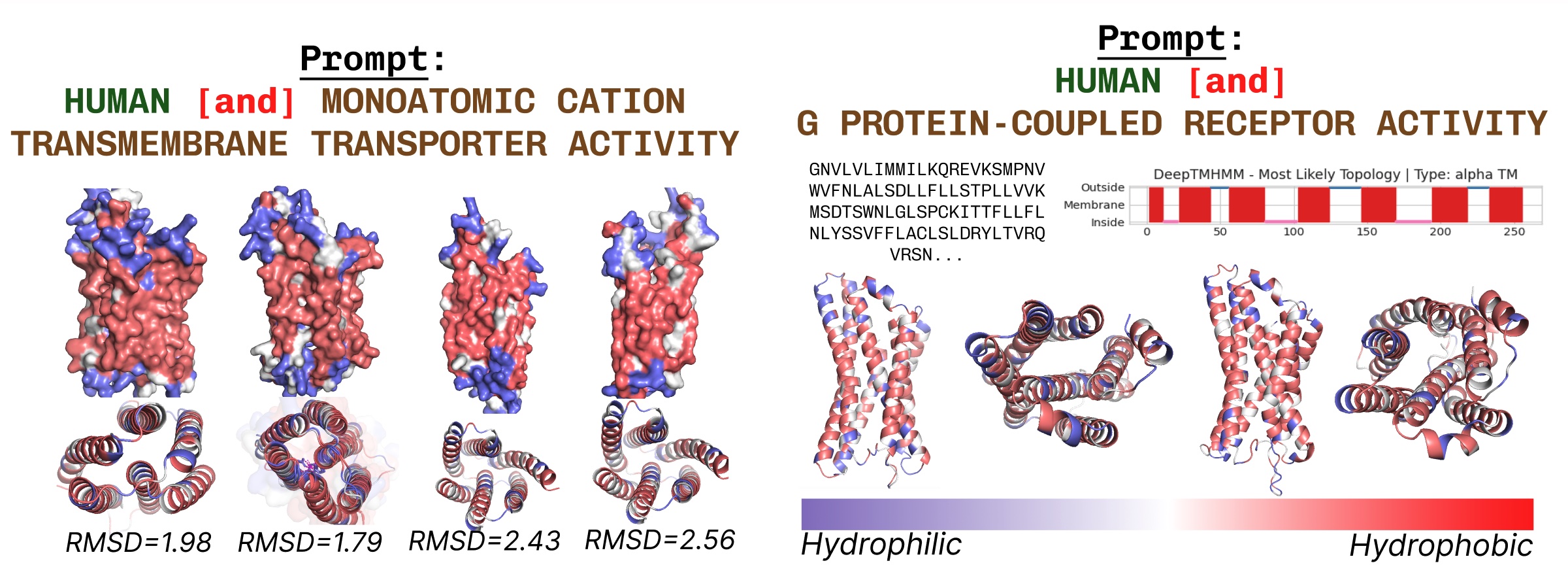
Las proteínas transmembrana tienen residuos hidrofóbicos en el núcleo, donde está incrustado dentro de la capa de ácidos grasos. Estos se observan consistentemente al solicitar el cuadrado con palabras clave de proteínas transmembrana.
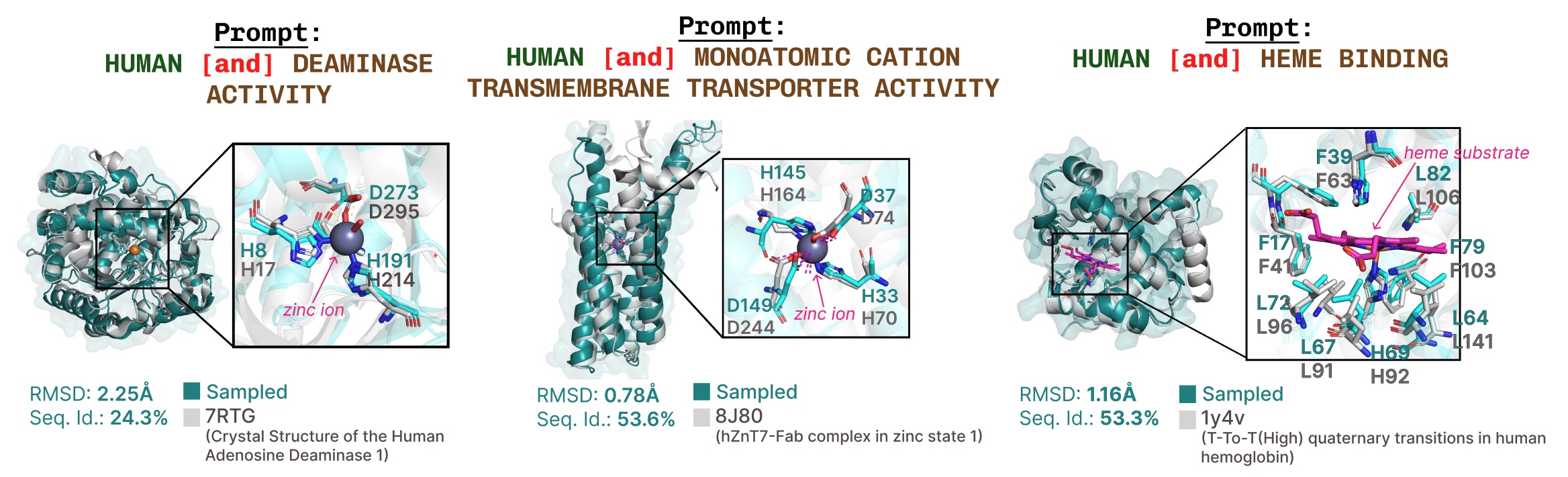
Ejemplos adicionales de recapitulación activa del sitio basada en la solicitud de palabras clave de función.
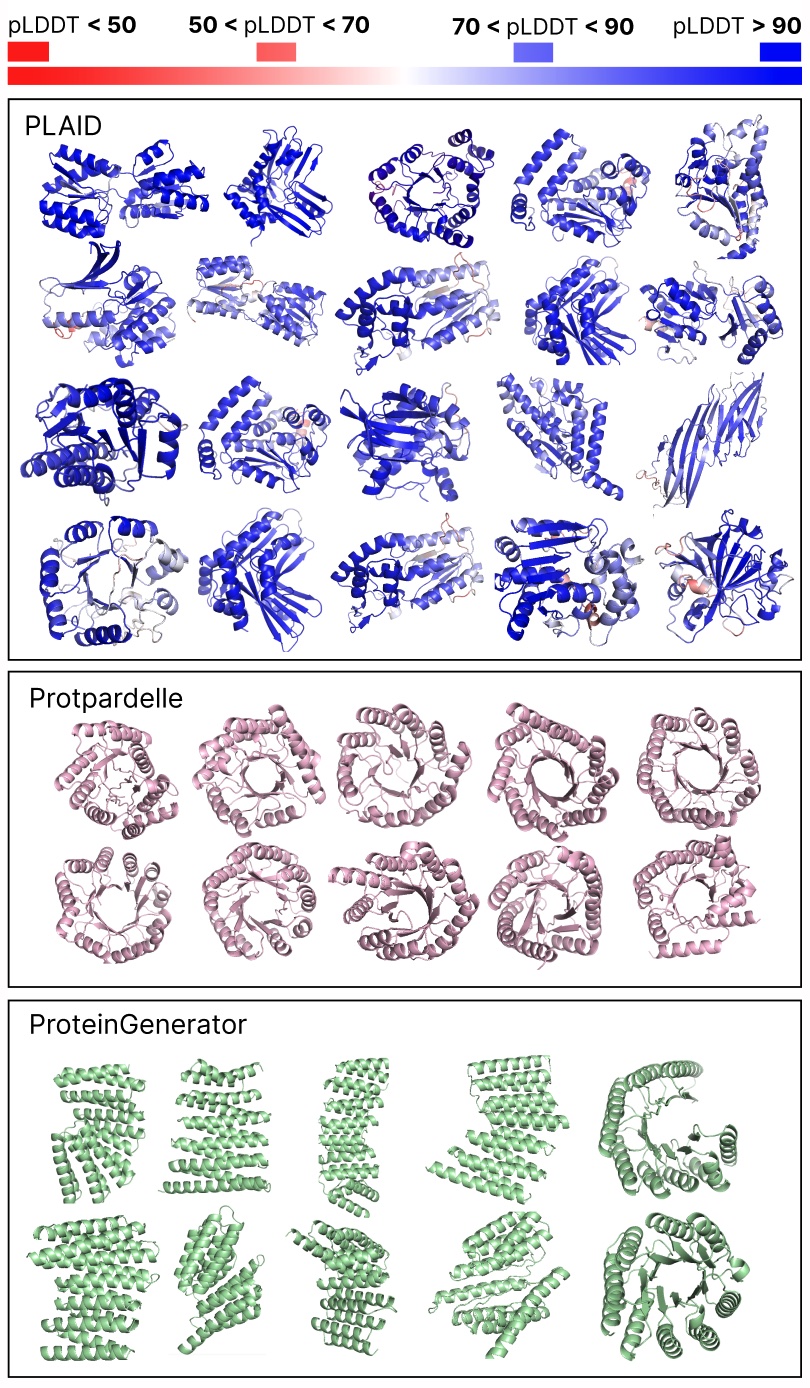
Comparación de muestras entre líneas de base a cuadros y totales. Las muestras a cuadros tienen una mejor diversidad y capturan el patrón de cadena beta que ha sido más difícil para los modelos generativos de proteínas.
Expresiones de gratitud
Gracias a Nathan Frey por sus comentarios detallados sobre este artículo, y a los coautores de Bair, Genentech, Microsoft Analysis y New York College: Wilson Yan, Sarah A. Robinson, Simon Kelow, Kevin Okay. Yang, Vladimir Gligorijevic, Kyunghyun Cho, Richard Bonneau, Pieterer, Abbeel y Nathan C. Freey.